







Malattia muscolare SMA
SMA sta per atrofia muscolare spinale. Questa malattia fa sì che i muscoli si indeboliscano gradualmente fino a non funzionare più. Prima si sviluppa la malattia, più velocemente i muscoli si deterioreranno. Solitamente i sintomi si manifestano nei bambini piccoli. Ogni anno a circa 10 bambini su 100.000 viene diagnosticata questa malattia. Per loro però arriva una buona notizia! Uno dei farmaci ha ottenuto l’approvazione dell’Agenzia Europea dei Medicinali, mentre un secondo farmaco è in fase di sperimentazione.
Cos’è la SMA?
La SMA è una malattia neuromuscolare: si tratta di un difetto nelle cellule nervose che trasmettono un segnale dal cervello ai muscoli. Queste cellule nervose sono chiamate “motoneuroni”. Se i muscoli non ricevono un segnale dai motoneuroni, non rispondono e quindi non si muovono. I muscoli che non fanno (quasi più) nulla diventano più deboli e sottili fino a non funzionare più. Alla fine i muscoli si paralizzano. La malattia muscolare SMA colpisce principalmente i muscoli delle braccia e delle gambe, ma con il progredire della malattia vengono interessati anche i muscoli del sistema respiratorio, provocando un’insufficienza di ossigeno nel corpo. Di conseguenza potrebbe essere necessaria una ventilazione cronica mediante l’aiuto di un dispositivo che migliora la respirazione.
La causa della SMA
La causa della SMA è un difetto in uno dei circa 20.000 geni che gli esseri umani possiedono. Si tratta di frammenti di DNA che determinano l’aspetto di un individuo, come è e se è sano. I geni si trovano sui cromosomi, che sono 46 per ogni persona (23 coppie). Gli scienziati hanno scoperto che la causa della malattia muscolare SMA si trova sul 5° cromosoma. Nei malati di SMA, il gene SMN1 è rotto o addirittura assente. Quello è il gene che normalmente produce la proteina SMN, che è il carburante per i motoneuroni. Senza questa proteina, i motoneuroni non possono funzionare (correttamente): i segnali provenienti dal cervello non vengono trasmessi ai muscoli correttamente o non vengono trasmessi affatto. Nei soggetti affetti da SMA, è il gene SMN2 (una sorta di gene “di riserva”) ad assumersi il compito di produrre la proteina SMN, ma produce solo il 10% della proteina utile che produce il gene SMN1.
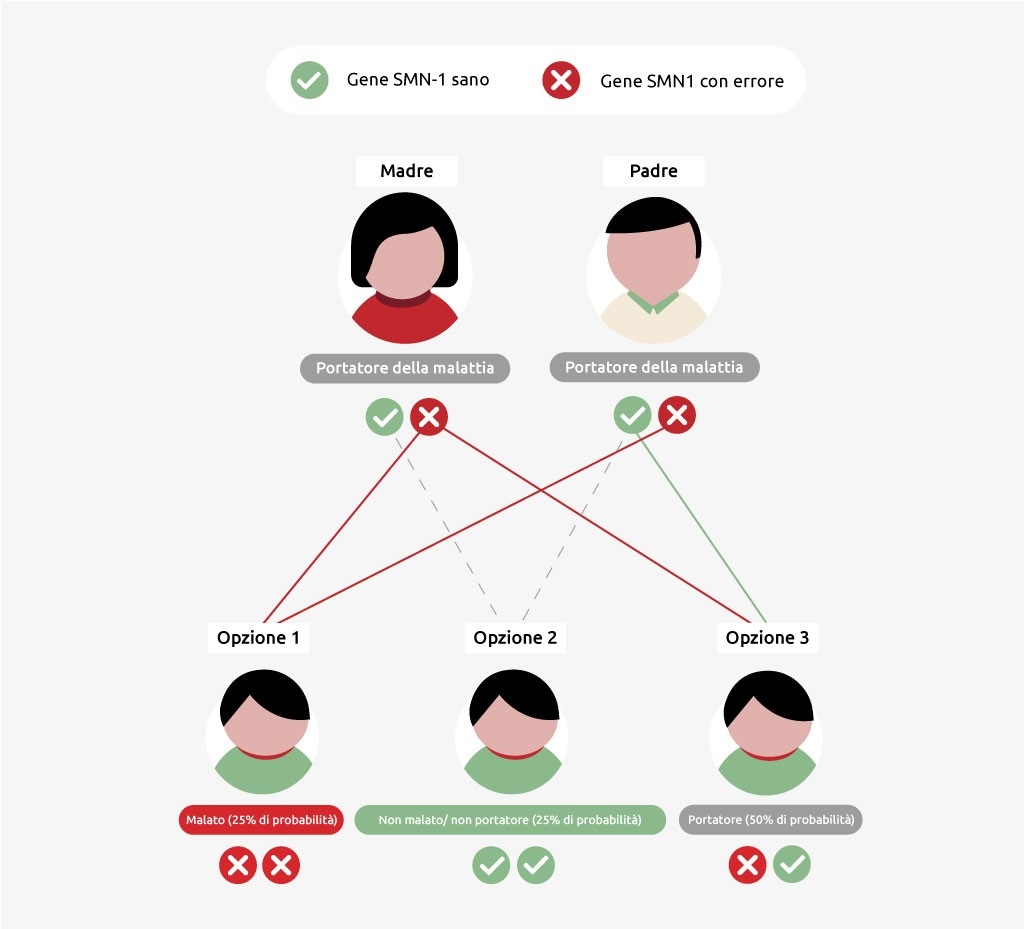
Ereditarietà della malattia SMA
La SMA è una malattia ereditaria: i bambini ricevono i geni affetti dai loro genitori. Se un genitore è portatore della malattia, ha un gene SMN1 sano e uno difettoso. Un portatore non è malato però può trasmettere la malattia. I bambini sviluppano la SMA solo se il padre e la madre trasmettono entrambi un gene SMN1 difettoso o un quinto cromosoma sprovvisto di un gene SMN1. La probabilità che i figli di due portatori si ammalino è del 25%. La possibilità che i bambini diventino soltanto portatori è più alta: 50%. Hanno anche il 25% di probabilità di non ereditare la malattia o di non essere affatto portatori.
I 4 tipi di SMA
I tipi di atrofia muscolare spinale sono 4. La distinzione è determinata dall’età della comparsa e dalla rapidità con cui i sintomi peggiorano. Poiché la malattia è spesso diagnosticata in giovane età, la SMA può essere considerata una malattia dei neonati e dei bambini piccoli. Più precoce è la comparsa della malattia, più grave è il suo decorso.
La differenza nel decorso della malattia è dovuta al fatto che alcune persone hanno più geni “di riserva” del gene SMN2. Quanti più di questi geni una persona presenta, tanto più la proteina SMN potrà essere prodotta e quindi meno mancherà ad una persona. Ciò permette ai motoneuroni di lavorare più a lungo. Un individuo con 0-2 copie sviluppa il tipo 0 o 1. Con 3 copie, si sviluppa il tipo 1, il tipo 2 o anche il tipo 3. Con 4 o più copie, si sviluppa il tipo 2 (in alcuni casi), il tipo 3 o il tipo 4.
Malattia muscolare SMA tipo 1
Il tipo 1 della SMA si verifica nei bambini, poco dopo la nascita o nei primi sei mesi. I muscoli della parte superiore delle braccia, delle gambe e del tronco sono spesso indeboliti. Anche i muscoli respiratori si indeboliscono. I bambini con la SMA di solito non imparano a stare seduti, a tenere la testa dritta o a girarsi. Questo tipo rappresenta la forma più grave della malattia. Anche in questo caso, prima si manifesta la malattia, peggiori saranno i disturbi. In alcuni casi la malattia può manifestarsi prima della nascita; si tratta della SMA tipo 0. Il tipo 1 si verifica in circa il 55% dei soggetti affetti da SMA.
Malattia muscolare SMA tipo 2
Il tipo 2 della SMA si manifesta nei bambini tra i 6 e i 18 mesi. Soffrono prevalentemente di muscoli indeboliti nelle gambe e nella schiena. Le braccia sono spesso ancora piuttosto forti. I bambini con questa forma di SMA possono imparare a sedersi e girarsi, ma non sono in grado di camminare autonomamente. I bambini e gli adolescenti con il tipo 2 hanno quindi quasi sempre bisogno di una sedia a rotelle. L’aspettativa di vita per la SMA tipo 2 è tra i 10 e i 40 anni. Può quindi variare molto da persona a persona.
Malattia muscolare SMA tipo 3
Quando la malattia muscolare SMA si sviluppa in bambini tra i 18 mesi e i 4 anni di età, si parla del tipo 3. I bambini imparano spesso a sedersi e a camminare autonomamente. Tuttavia, c’è una buona probabilità che perdano queste abilità più tardi perché i loro muscoli diventeranno più deboli. I bambini con la SMA 3 hanno quindi spesso bisogno di usare una sedia a rotelle. In una fase successiva, potrebbe essere necessario utilizzare un respiratore. Anche con il tipo 3 l’aspettativa di vita varia. Le persone a cui è stata diagnosticata la SMA di tipo 3 di solito hanno un’aspettativa di vita normale.

Malattia muscolare SMA tipo 4
Il tipo 4 della SMA è piuttosto raro. In questo tipo, i sintomi non appaiono se non dopo i 30 anni. La maggior parte delle persone con questo tipo accusano solo sintomi lievi. Fasi di peggioramento e periodi senza sintomi si alternano. Gli individui con la SMA tipo 4 hanno spesso un’aspettativa di vita normale. Tuttavia, in tarda età, potrebbero avere difficoltà a fare le scale e a sollevare le braccia.
Effettuare la diagnosi
Nella maggior parte dei casi, l’atrofia muscolare spinale viene diagnosticata attraverso il test del DNA. Tale test mostra la presenza di eventuali anomalie nel DNA ma non è possibile determinarne la tipologia. Ciò dipende da quando ha inizio la malattia e da quanto velocemente si sviluppa.
Nel restante 8%, non è possibile identificare l’anomalia del gene. Allora è necessario un altro tipo di esame: l’esame elettromiografico (EMG) o una biopsia muscolare. Nell’esame elettromiografico vengono inseriti nei muscoli degli aghi sottili. Questi sono collegati al dispositivo EMG che misura le reazioni dei muscoli. Il medico può quindi vedere se i muscoli reagiscono in modo anomalo. In una biopsia muscolare il dottore rimuove un pezzettino di muscolo per studiarlo al microscopio, ne esamina la struttura ed esegue dei test. Osserva, ad esempio, la dimensione delle fibre muscolari. Se molto diverse, la malattia del muscolo SMA potrà essere constatata.
Cura della malattia
Sono stati sviluppati diversi farmaci per trattare la SMA e ridurne i sintomi. Al momento, in Europa è stato approvato il farmaco Spinraza. Attraverso un’epidurale, il gene SMN2 viene influenzato più volte all’anno in modo tale che possa produrre più proteina SMN impiegabile. Ciò fornisce ai motoneuroni più carburante per controllare i muscoli.
Esiste anche un farmaco chiamato Zolgensma, che utilizza la terapia genica per creare un nuovo gene SMN1 nei motoneuroni. Il vantaggio è che questo farmaco deve essere usato una sola volta. Gli svantaggi sono il costo di 2 milioni di euro per paziente e l’incertezza degli effetti a lungo termine. In Europa, il farmaco è stato approvato per l’uso nei bambini piccoli. Tuttavia è necessario prendere accordi sul rimborso del farmaco prima che questo possa essere effettivamente utilizzato.
Di più al riguardo salute